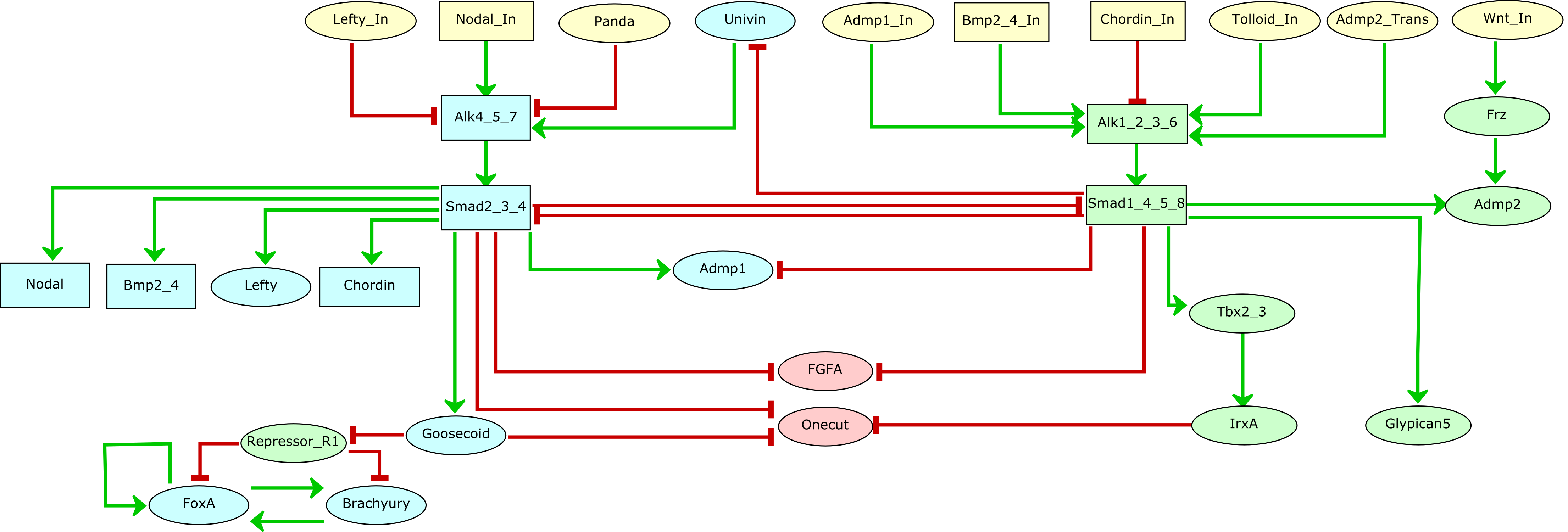
Drosophila DPP (TGF-beta homolog) signalling pathway is triggered by ligand-induced formation of heterotetrameric complexes consisting of two type II receptors and two type I receptors with intrinsic serine/threonine kinase activity. The type I receptor (SAX or TKV) is phosphorylated by the constitutively active type II receptor kinase (Punt). Consequently, the complex becomes active and phosphorylates the receptor-regulated Smads (R-Smads). Phosphorylated R-Smads (MAD and Smox) form complexes with a common-mediator Smad (Medea) and translocate into the nucleus, where they regulate the transcription of target genes in co-operation with other transcription factors (nejire, schnurri). DPP is a morphogen, i.e. a molecule distributed in a concentration gradient that elicits different cell fates as a function of its concentration, thereby organizing a series of cell types in a defined spatial array. In response to DPP gradient, cells adopt different fates. The establishment of dpp gradient involves the proteins SOG and TSG. These proteins together capture the DPP ligand and prevent its binding to the receptor (Punt). The heteromeric complex (SOG, DPP, TSG) then release the DPP ligand, a process involving the cleavage of SOG by Tolloid (a metalloprotease). Other TGF-beta signals, Glass-bottom-boa (GBB) and Screw (SCW), help DPP to potentiate cells to respond. SCW and GBB are never expressed together in the same region and affect different cells during: i) early D/V patterning of the embryo and specification/differentiation of dorsal cells (if there is no screw, dpp alone is unable to establish the D/V pattern and embryo lack amnioserosa); ii) the development of adult structures such as the wing. GBB or SCW form heterodimeric complexes with DPP. These heterodimers can only signal through TKV, while SCW/SCW and GBB/GBB signals trough SAX, and DPP/DPP trough TKV and SAX. To model DPP signalling and the formation of the gradient, we have considered three different levels for the TKV receptor (0, 1, 2) and the MADMED effector (0, 1, 2). The regulatory graph also accounts for the potentiation of responding cells due to association of DPP and SCW, or of DPP and GBB. Activated by MADMED, DAD is a pathway inhibitor that can modulate the pathway activity from high to low signalling. DAD works by abrogating the phosphorylation of the MADMED complex by TKV or SAX, thus involving a negative circuit between DAD and the MADMED complex. In addition, BRK another inhibitor of the DPP pathway can block the transcription of dad. Our model reproduces the formation of the DPP signalling gradient and accounts for the role of the heterodimers signalling in cell potentiation. To simulate DPP signalling, we start from an initial state corresponding to non differentiated cell, that can receives high or low level of DPP signal. The use of ternary nodes enables us to account for differential effects of different DPP levels (gradient). The cells receiving high level expression display the hetero-dimers SCW/DPP or GBB/DPP and correspond to Tld expression area, which promotes DPP gradient formation. In presence of medium DPP, TSG and SOG but no TLD are initially needed to capture homo- or hetero-dimer, diminishing pathway signalling intensity (expression level 1 for TKV and MADMED). In presence of high pathway signalling, two situations occur: i) in cells potentiated by SCW: a sequestering complex (SOG/TSG/ DPP/SCW) will release the signalling molecule upon TLD clivage, in addition to normal DPP signalling. This leads to a higher signal transduction. ii) in cells potentiated by GBB, the situation is similar but involve a different heterodimer (GBB/DPP). These situations correspond to two different stable states with high TKV and MADMED (level 2), denoting that more receptors are required to enable a higher level of nuclear MADMED. We consider five different initial states: i) the first one corresponds to the absence of signalling, i.e. absence of DPP; ii) the second one corresponds to medium signalling, characterized by the presence of Dpp at level 1 and of SCW; iii) the third one corresponds to medium signalling, characterized by the presence of Dpp at level 1 and of GBB); iv) the fourth one corresponds to the presence of DPP at level 2 and of SCW; v) the last one corresponds to the presence of DPP at level 2 and of GBB. These set of initial states enable the simulation of five situations. No signalling, two medium and two high signalling that characterize the behavior of the pathway. The stable state obtained with the no signalling simulation shows the absence of binding of the ligands to the receptors TKV and Punt (level of expression 0) and the non activation of target nodes. These medium signals simulations in presence of DPP, show the activation of the receptors (level of expression 1) and subsequent signalling cascade leading to the activation of pathway's targets. These medium signal are defined by the level of expression 1 for DPP, MADMED and TKV while in the high signalling sets, these nodes are expressed at level 2. The node Tkv is multi-valued because the high signalling is characterized by the binding of hetero dimers (DPP/SCW or DPP/GBB) signalling through TKV. Note that GBB and SCW don't have the same expression pattern. For more details on Dpp signalling pathway regulation see [1]; [2]; [3]; [4]; [5].
References
- Eldar A, Dorfman R, Weiss D, Ashe H, Shilo B-Z, Barkai N.
2002. Robustness of the BMP morphogen gradient in Drosophila embryonic patterning.. Nature. 419(6904):304-8.
- Kamiya Y, Miyazono K, Miyazawa K.
2008. Specificity of the inhibitory effects of Dad on TGF-beta family type I receptors, Thickveins, Saxophone, and Baboon in Drosophila.. FEBS letters. 582(17):2496-500.
- Wartlick O, Mumcu P, Kicheva A, Bittig T, Seum C, Jülicher F, González-Gaitán M.
2011. Dynamics of Dpp signaling and proliferation control.. Science (New York, N.Y.). 331(6021):1154-9.
- Baena-Lopez LA, Nojima H, Vincent J-P.
2012. Integration of morphogen signalling within the growth regulatory network.. Current opinion in cell biology. 24(2):166-72.
- Brooks A, Dou W, Yang X, Brosnan T, Pargett M, Raftery LA, Umulis DM.
2012. BMP signaling in wing development: A critical perspective on quantitative image analysis.. FEBS letters. 586(14):1942-52.